Disclaimer: I am not a licensed investment or medical professional, and the ideas herein are in no way intended as investment or medical advice. I do not have any personal investments in any of the companies mentioned at the time of this writing, nor do any of my employers (to the best of my knowledge). The sole purpose of this write-up is to share my personal perspective and encourage others to share their thoughts on the future of DMD therapy.
Introduction
Dystrophin is a rod-shaped protein that links actin filaments to the muscle cell membrane. Mutations that reduce functional dystrophin levels cause Duchenne muscular dystrophy (DMD), a progressive muscular disease. The average life expectancy in DMD is 26 years. The standard of care (SOC) corticosteroid (CS) therapy stimulates muscle regeneration in young patients and reduces inflammation. However, long-term use of CS drugs is associated with severe side effects (e.g., Cushingoid appearance) and can only delay disease progression by 2-3 years. Given DMD biology, restoring the expression of functional dystrophin is necessary to prevent continued patient decline. The following technologies have been applied to the problem:
- Antisense Oligonucleotide (AO)-mediated skipping of mutated exons
- Small molecule-mediated ribosomal readthrough of nonsense mutations
- AAV-mediated transfer of microdystrophin, a shortened DMD transgene
Several drugs from the first category have been conditionally approved based on inducing dystrophin expression but failed to demonstrate functional benefit, as measured by the 6-Minute Walk Test (6MWT). In the second category, one drug was conditionally approved based on a borderline significant 6MWT improvement seen in post hoc analysis. None of these recently approved drugs surpassed the SOC on functional benefits in a randomized, prospective study.
The third category consists of three gene therapy programs: PF-06939926 (Pfizer), SRP-9001 (Sarepta Therapeutics), and SGT-001 (Solid Biosciences). So far, gene therapies surpassed other treatment modalities in increasing dystrophin levels. That said, Sarepta is now approaching similar levels with its second-generation AO drug, SRP-5051. Importantly, gene therapies also demonstrate their potential on functional endpoints, while clinical benefit data on SRP-5051 has not yet matured.
Between the three gene therapy programs and the second-generation AO, SRP-5051, I selected SGT-001 (Solid Biosciences) and SRP-5051 (Sarepta) as the two most promising therapeutic agents. My selection was based on four major factors: efficacy, mechanism of action, biodistribution, and safety.
Efficacy
Feraudy et al. (2021) found that residual dystrophin levels as low as <0.5% of normal could shift DMD to the milder Becker muscular dystrophy (BMD) phenotype. Approved AO drugs increase dystrophin expression up to 5% of normal. However, like the ribosomal readthrough drug, ataluren, they fail to demonstrate statistically significant clinical benefit, as measured by 6MWT (Table 1).
Table 1. Efficacy and safety of targeted DMD therapy

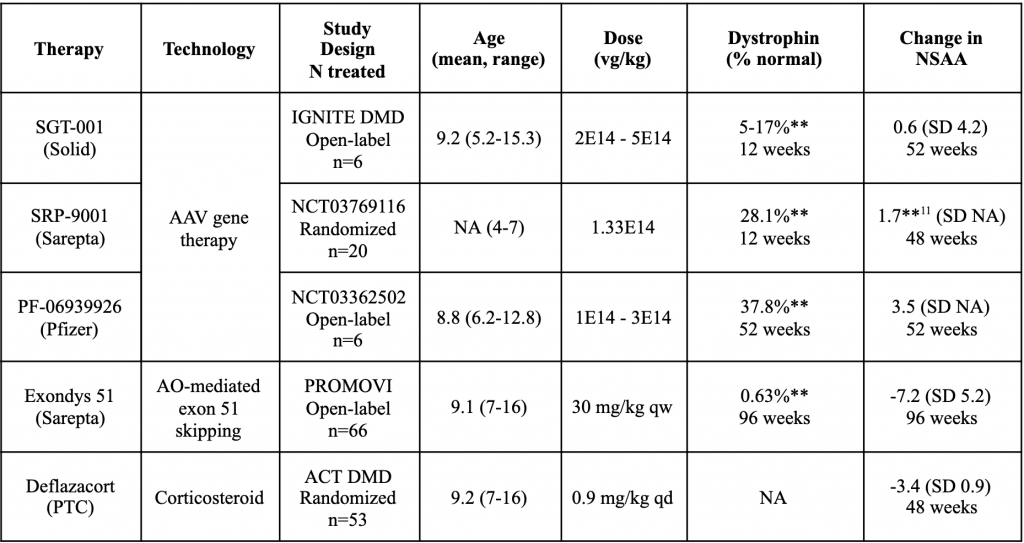
AAV gene therapies surpassed other DMD treatments in increasing dystrophin tissue levels. At most, an AO drug (SRP-5051) raised dystrophin expression to 6.5% of normal after 12 weeks (Table 1). In contrast, AAV gene therapies achieved 10-40% (Table 2). More importantly, gene therapies demonstrated previously unseen efficacy potential on functional endpoints. Notably, Sarepta’s gene therapy, SRP-9001, showed a statistically significant North Star Ambulatory Assessment (NSAA) increase over baseline in the treatment arm of a randomized study (n=20). Gene therapies from Pfizer and Solid also showed an NSAA effect in small open-label trials (n=6).
Table 2. Comparative efficacy of AAV gene therapies vs. the SOC
Solid’s gene therapy, SGT-001, also improved 6MWT and FVC% functional outcomes (Table 3).
Table 3. Comparative efficacy of SGT-001 gene therapy on FVC% and 6MWT measures
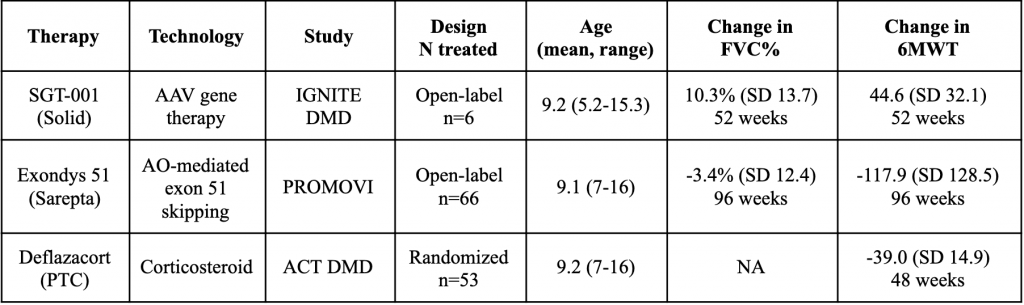
FVC% is closely linked to the survival of DMD patients and has a history of being used as a registrational endpoint in other progressive myopathies. Thus, while it is premature to judge the relative clinical efficacy of AAV gene therapies and SRP-5051 until more randomized studies are performed, SGT-001 has demonstrated its potential on a broader set of functional endpoints.
Mechanism of action
DMD is one of the longest human genes and greatly exceeds the loading capacity of an AAV vector. An inspiration for microdystrophin came from a 61-year-old ambulatory patient with BMD, who had 46% of the dystrophin coding region deleted (England et al., 1990). Subsequent studies showed that in-frame deletions from the middle rod domain of dystrophin often lead to mild disease. The rod domain consists of 24 spectrin-like repeats (R1-R24) and 4 hinges (H1-H4). Different parts of the rod domain are removed in the three AAV-delivered transgenes, so the resulting proteins differ in the remaining hinges and repeats (Table 4).
Table 4. Structural features of microdystrophin transgenes
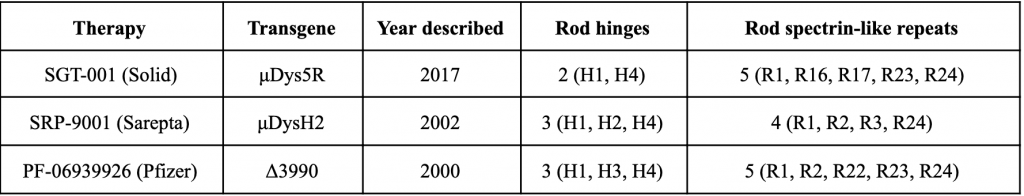
The μDys5R transgene in SGT-001 is distinct in keeping the repeats R16-R17 that comprise the nNOS-binding domain. nNOS stimulates vasodilation in response to exercise. BMD patients often have reduced nNOS localization to the muscle cell membrane (sarcolemma) and exhibit profound muscle fatigue even after a mild exercise (Kobayashi et al., 2008). Importantly, SGT-001 trial showed co-localization of nNOS and microdystrophin at the sarcolemma of treated patients.
Sarepta’s μDysH2 transgene keeps the H2 hinge. Banks et al. (2010) demonstrated that a conserved polyproline site in H2 disrupts normal muscle structure in mdx mice. Even though Sarepta’s microdystrophin has not raised such issues in a human trial, its pathogenic action in the mouse DMD model is concerning.
Pfizer’s Δ3990 transgene keeps the H3 hinge. Evidence regarding the benefits of including H3 is mixed. One study of DMD patients found that in-frame deletions of H3 yielded milder disease (Carsana et al., 2005). However, another study found that including H3 was more effective in lessening muscle pathology in mdx mice (Harper et al., 2002).
In summary, Solid has the best-in-class microdystrophin due to the presence of the nNOS-binding domain. Its value was also validated by Ultragenyx that paid $40M upfront to license Solid’s construct for its gene therapy based on another AAV variant. That said, microdystrophin is significantly shorter than wild-type dystrophin (167 vs. 427 kDa). In contrast, exon 51 skipping induced by SRP-5051 results in a 420 kDa protein. Thus, Solid’s transgene is likely to be more functional than other microdystrophins, but nearly full-length dystrophin induced by Sarepta’s second-generation AO is probably better still. Based on the molecular features of resulting dystrophins, SGT-001 (Solid) and SRP-5051 (Sarepta) are the top choices within their respective drug classes.
Biodistribution
Heart failure is the leading cause of DMD mortality, thus delaying or stopping the cardio progression of DMD is essential to improving survival. All three AAV gene therapies use different promoters to drive tissue-specific expression of their transgenes. The CK8 and MHCK7 promoters used by Solid and Sarepta drive expression in both skeletal and cardiac muscle. However, Pfizer uses a skeletal muscle promoter that does not drive any expression in the heart (Meyers et al., 2019). This is a critical drawback of Pfizer’s gene therapy.
Poor cardiac uptake is also a major downside of first-generation AO drugs based on PMO chemistry (e.g., Exondys 51, Viltepso). However, SRP-5051 is based on the PPMO platform that adds a charged peptide designed to improve cellular uptake. In nonclinical studies, SRP-5051 induced robust cardiac expression in mice (Hadcock et al., 2021) and nonhuman primates (Gan et al., 2019). Thus, SRP-5051 (Sarepta) and SGT-001 (Solid) are superior to Pfizer’s gene therapy in having the potential to address a critical unmet need: stopping the cardio progression of DMD.
Safety
AAV9-based therapies (SGT-001 and Pfizer’s PF-06939926) exhibited a high frequency of complement activation SAEs, such as thrombocytopenia and acute kidney injury. Sarepta’s SRP-5051 caused grade 4 hypomagnesemia (Table 5).
Table 5. Comparative safety of AAV gene therapies and SRP-5051

Following a clinical hold, Solid Biosciences revised its manufacturing to remove the majority of empty viral capsids and reduce the total viral load of SGT-001 (Table 5). It also modified its trial protocol to include pretreatment with complement inhibitors, safely dosing the next patient in a resumed trial. Despite these safeguards, any possibility of complement activation may still seem excessive compared to grade 1-2 side effects of AO and CS therapy (Table 1). However, AAV gene therapy is a one-time treatment, and its long-term impact on patient survival and quality of life may eclipse its short-term risks.
The hypomagnesemia side effect of SRP-5051 is thought to be unrelated to renal function and caused by the drug’s interference with magnesium transporters. While severe, it proved to be transient and reversible with oral magnesium supplementation. Thus, SRP-5051 and SGT-001 have serious but manageable side-effects, balanced by their efficacy potential.
Conclusion
Solid’s AAV gene therapy (SGT-001) and Sarepta’s second-generation AO (SRP-5051) demonstrate the best balance between safety and efficacy within their respective drug classes. How do they compare on market positioning and pricing?
On pricing, SGT-001 is likely to be valued higher than any chronic therapy. As seen in SMA, a one-time gene therapy treatment can secure a much higher price point (Zolgenzma, $2.1M) than a periodic oligonucleotide injection (Spinraza, $375K). That said, one should appreciate the added convenience of SRP-5051 monthly dosing and the price premium it could earn, compared to the weekly regimen of the first-generation AO therapy (Exondys 51, $750K).
Most importantly, SGT-001 and SRP-5051 are likely to occupy different market niches. SRP-5051 might be a better option for older, non-ambulatory patients, amenable to exon 51 skipping. SGT-001 might be well-suited for young, ambulatory patients without neutralizing antibodies to AAV9. Finally, in some patients, these drugs may be combined with the one-time SGT-001 treatment followed by chronic therapy with SRP-5051.